Los pacientes con SCdL presentan un fenotipo característico en el que destaca una facies peculiar, alteraciones en las extremidades y retraso del desarrollo pre y postnatal con retraso psicomotor/mental. Además, presentan malformaciones congénitas que afectan a distintos órganos o sistemas, que a continuación desarrollamos.
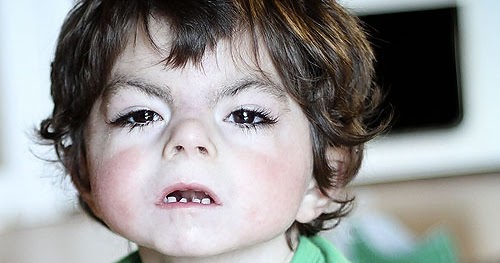
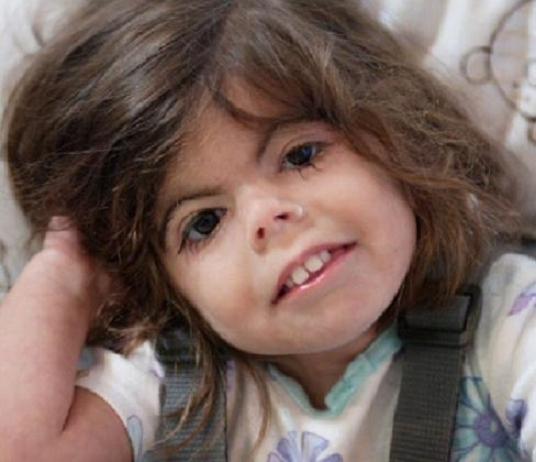
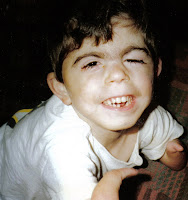
Los rasgos craneofaciales en el SCdL incluyen microcefalia, sinofridia con cejas arqueadas, pestañas largas y finas, nariz pequeña con puente nasal deprimido y ancho, narinas antevertidas y un filtrum alargado y prominente. Presentan además un labio superior fino con comisuras orientadas hacia abajo, paladar elevado, diastema dentario y micrognatia. Los pabellones auriculares son de implantación baja y rotados hacia atrás. El seguimiento de los individuos con sospecha de SCdL es importante ya que algunos hallazgos clínicos pueden pasar inicialmente desapercibidos.
Estos pacientes presentan un hirsutismo generalizado, que se aprecia sobretodo en la cara, espalda y extremidades. Además, suelen tener un cuello corto con una implantación baja de la línea posterior del cabello.